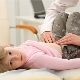
Spinaalinen lihasten atrofia lapsilla

Selkärangan lihasten atrofia on vakava patologia, joka tekee usein yksinkertaisia toimia, kuten lapsen kävelemistä, istumista. Vauvasta voi jäädä jopa niin luonnollinen tilaisuus itsenäisenä hengityksenä. On hyvin vaikeaa tehdä ennusteita tästä patologiasta, loppujen lopuksi ei ole olemassa erityistä hoitoa eikä ennaltaehkäisyä, mutta kaikki riippuu sairauden muodosta ja tekijöistä, mikä lääke ei löydä selitystä.
Mikä tämä on?
Spinaalisen lihaksen atrofian puhuminen ei tarkoita yhtä tiettyä sairautta, vaan koko sairauksien yleistä lyhennettä CMA. Kaikki ne ovat perinnöllisiä ja liittyvät selkäytimen hermosolujen rappeutumiseen, jotka ovat vastuussa moottorin toiminnoista.
Niistä geneettiset patologiat lasten selkärangan lihasten atrofioissa ovat levinneisyyden suhteen johtavia paikkoja. Ja noin joka kuudes tuhatta lasta syntyy niin kauhean diagnoosin avulla. 50 prosentissa tapauksista lapset eivät asu kahden vuoden iässä ja kuolevat. Loput elämästä on vamma.

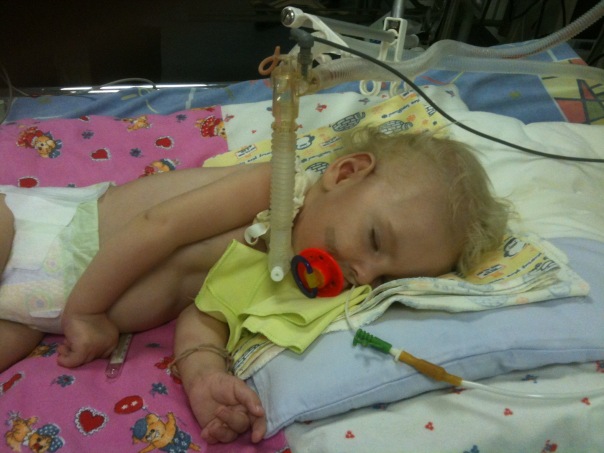
Geneettisten asiantuntijoiden mukaan ongelma on paljon laajempi kuin näyttää siltä, että tilastot ovat.
Sairaus kehittyy tiettyjen geenien mutaation vuoksi, ja yksi niistä on SMN1, joka pidetään patologian tärkeimpänä "syyllisenä", muunnellussa muodossa, joka esiintyy taantumattoman läsnäolon muodossa planeetan jokaisessa viisikymmentä asukasta. Tämä tarkoittaa, että terveillä vanhemmilla, jotka eivät edes ymmärrä, että ne ovat reseptorisia mutantin geenin kantajia, voi olla vauva, jolla on selkärangan lihasten atrofia.
Sairauksien ryhmää kuvattiin ensin 1800-luvulla Guido Verding, jonka nimi oli myöhemmin nimetty yhdeksi AGR: n lasten lajikkeista.
luokitus
Yleisin SMA-muoto lapsilla on proksimaalinen. Sitä edustavat useat sairaudet, joista kaikki eivät ilmene heti lapsen syntymän jälkeen.
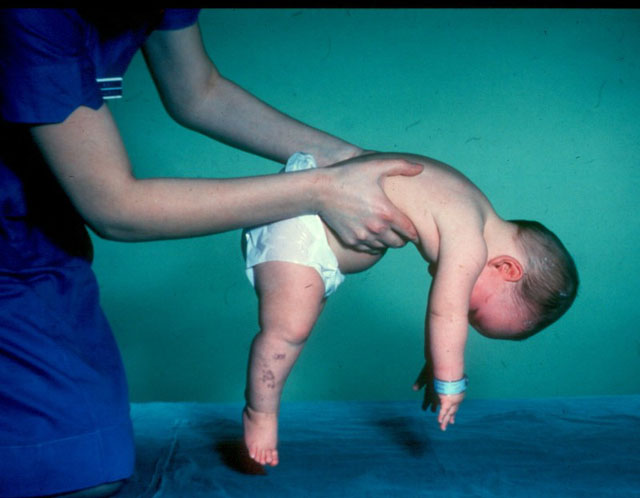
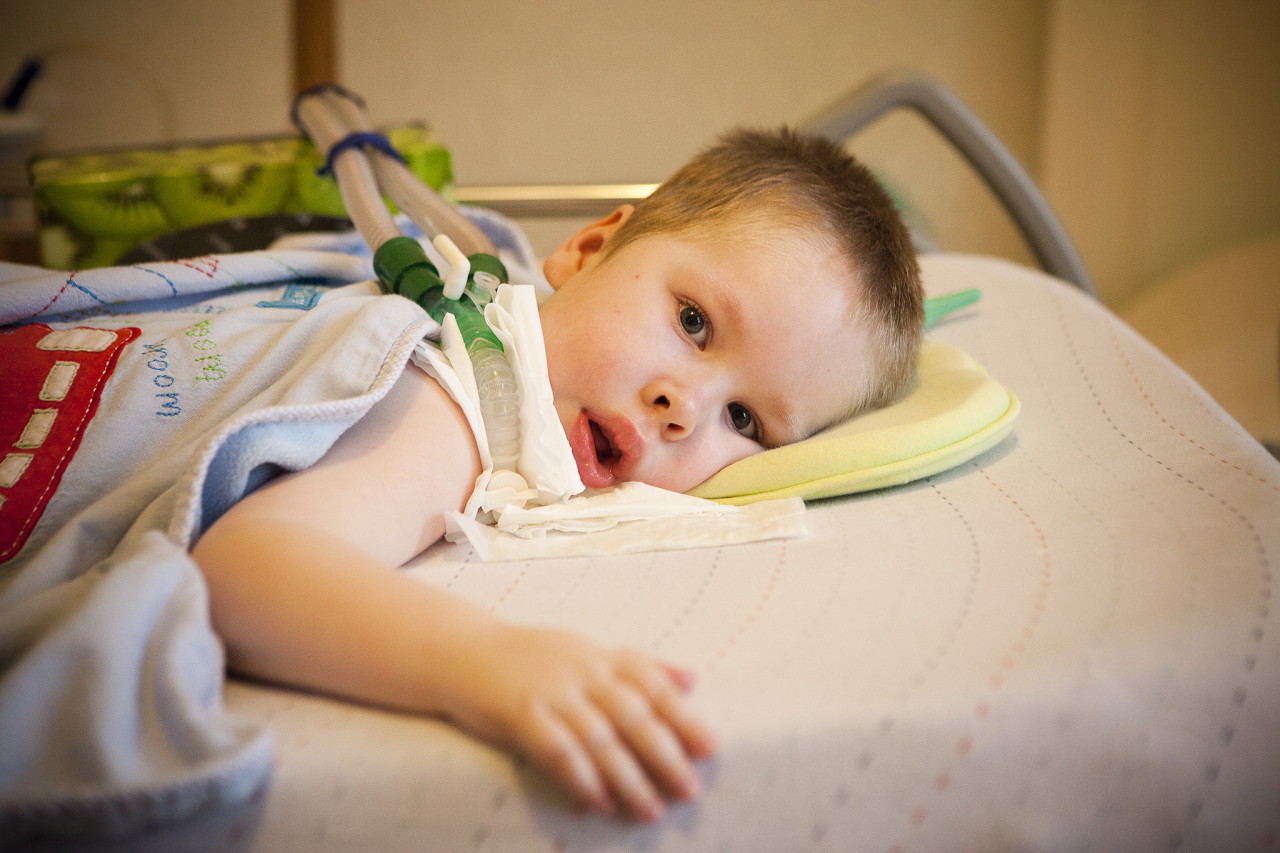
- Verding-Hoffman-tauti - tyyppi 1 MCA, vakava imeväisairausjoka ilmenee lapsen elämän kuuden ensimmäisen kuukauden aikana. Eniten ennustetaan hänen epäsuotuisimpia, useimmat potilaat kuolevat. Lapsi, jolla on tyyppi 1 SMA, ei voi seisoa, istua eikä vierittää itsenäisesti. Monilla vastasyntyneillä on heikentynyt imu- ja nielemisrefleksit. Usein ei ole mahdollista spontaania hengitystä tai hengitys on vaikeaa.
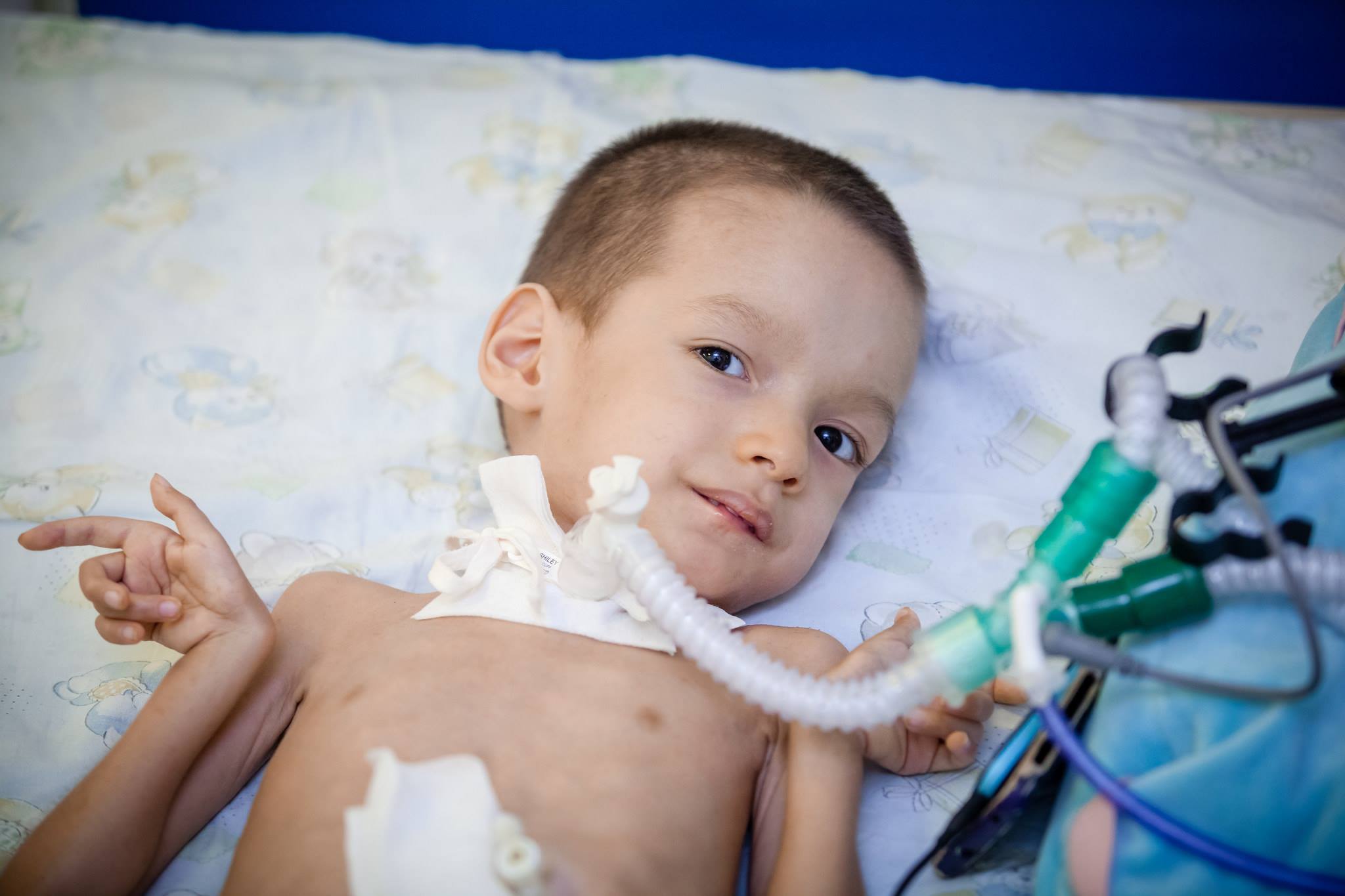
- Atrofia Dubovitsa - CMA tyyppi 2, myöhäinen lapsi. Se tapahtuu yleensä kuuden kuukauden ja puolen vuoden välein. Lapsi ei voi kävellä, seistä, mutta pystyy istumaan, ruoka ei häiritse, hän selviytyy nielemisen tehtävistä, imee. Kuinka kauan vauva elää riippuu hengityselinten lihasten tilasta.
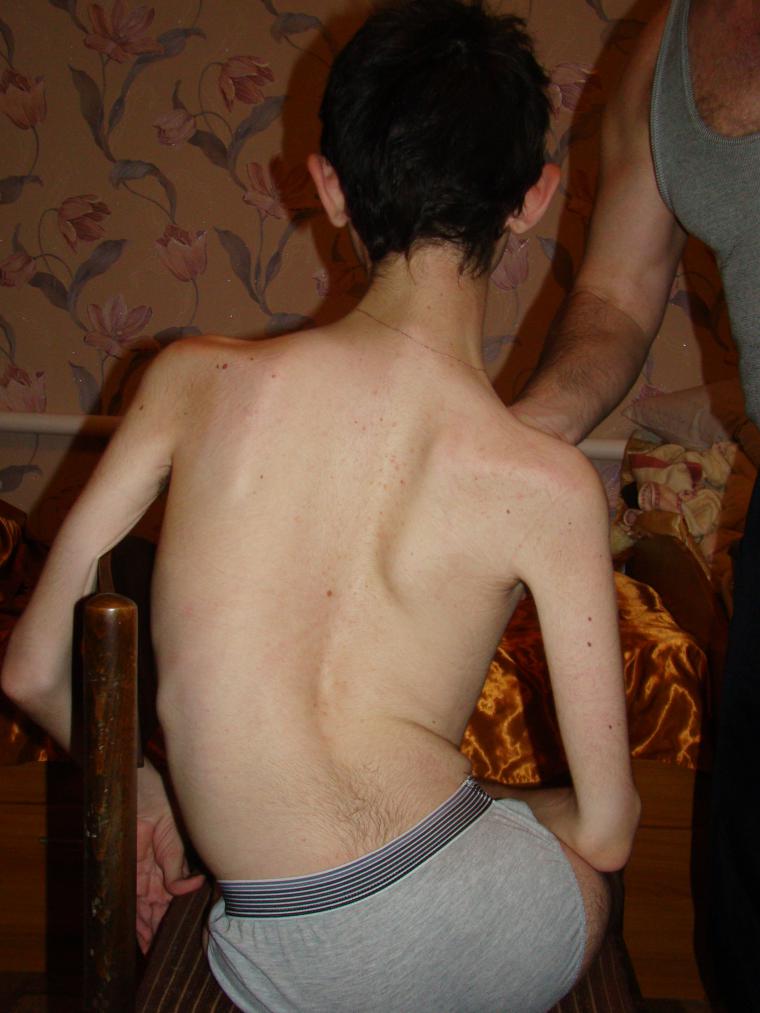
- Kugelberg-Welanderin - CMA 3 -tyyppien atrofia, infantiili. Yleensä löytyy puolitoista vuotta, yleensä kahden vuoden kuluessa. Prostostisesti edullisempi muoto. Pienet potilaat voivat seistä, istua, liikkua, mutta kokea suurta heikkoutta, ja siksi useimmissa tapauksissa tarvitsevat pyörätuolin, jota ilman heidän normaalia elintärkeää toimintaa on heille vaikea.
- Kennedyn atrofia - CMA-tyyppi 4, bulbospinalnaya. Yleensä katsotaan aikuisen muotoon, mutta joskus havaitaan lapsilla 15 vuoden kuluttua. Elämän vaikutus vaikuttaa harvoin, lihasten heikkeneminen tapahtuu hitaasti, vähitellen, henkilö, joka johti normaalia elämää ja katsoi olevansa terve, lopulta vammautuu ja menettää kykynsä liikkua itsenäisesti.


Enemmän tai vähemmän tunnettua Duchennen ensimmäistä käyntiä ja Vulpianin atrofiaa on SMA ”aikuiset”, ensimmäinen havaitaan yleensä 18 vuoden jälkeen ja toinen 20 vuoden kuluttua.
Lapsissa ei tallenneta vain yksittäisiä SMA-muotoja, kun mikään ei häiritse lihasten dynaamisuutta, mutta myös yhdistettyjä muotoja, kun selkärangan atrofia ei ole ainoa diagnoosi ja lapsella on muita geneettisiä tai synnynnäisiä ongelmia, kuten sydän- ja verisuonivirheet, oligofrenia.
syistä
Kuten jo mainittiin, puhumme geneettisestä sairaudesta, ja siksi sen esiintymisen syyt ovat geneettikoiden etsintäalue. Lapsi perii yhden resessiivisistä geeneistä viidennellä kromosomilla (nämä voivat olla SMN, NAIP, H4F5, BTF2p44-geenit).
Todennäköisyys siirtää tällainen geeni kantajalle jälkeläisille on korkea - 25%. Jos sekä äiti että isä ovat mutaattisen geenin piilotettuja kantajia, niin AGR: n todennäköisyys lapsessa on 50%. Vaikuttavat epänormaalit geenit estävät normaalin SMN-proteiinin tuotannon ja selkäydin lihasten moottoritoiminnoista vastaavat hermosolut kuolevat vähitellen. Heidän kuolemansa prosessi jatkuu vauvan syntymän jälkeen.


ilmenemismuotoja
Oireet riippuvat sairauden tyypistä. Koska pidämme vain neljän tyyppisiä lapsia, on huomattava, että lihaksen heikkous ja lihasten atrofia ovat kaikille ominaisia. Muussa tapauksessa jokaisella tyypillä on oma kliininen kuva ja erottamiskyky.
- CMA-tyyppi 1 (Verding-Hoffman-atrofia) saatavilla myös raskauden aikana. Lääkäri saattaa epäillä sikiön sairautta hyvin hitaasti sekoittaen. Mutta vahvistaakseen diagnoosin lapsen kuljettamisen vaiheessa on vaikeaa, se tapahtuu yleensä synnytyksen jälkeen. Lapsi, jolla on tällainen atrofia, ei voi pitää päänsä itseään, heillä ei voi istua. Hän melkein aina makaa selässä, hänen asennonsa on rento, hän ei nosta jalkojaan, ei tuo heitä yhteen, ei laita kämmentensä yhteen. Hyvin varhaisessa vaiheessa voi olla suuria ongelmia lapsen ruokkimiseksi, koska hän nielee sen osoittautuu erittäin huonoksi tai ei toimi. Suurin osa lapsista kuolee ennen kahden vuoden ikää. Jotkut onnistuvat elämään seitsemän tai kahdeksan vuoden ajan, mutta atrofia vain pahenee. Yleensä kuolema johtuu sydämen, keuhkojen, ruoansulatuselinten vajaatoiminnasta.
- CMA 2 -tyyppi (atrofia Dubovitsa) syntymähetkellä sitä ei yleensä havaita, koska lapsi voi hengittää, niellä ruokaa, ja vasta kuuden kuukauden kuluttua ilmenee lihas atrofian eteneminen. Jos ensimmäiset oireet ilmenevät iässä, jolloin lapsi on jo oppinut seisomaan sängyn pinnassa, sitten leikkaussilmukka jaloista, kohtuuton murusien pudotus voi olla kirkas merkki. Vähitellen on vaikeaa niellä. Ajan myötä lapsi alkaa tarvita pyörätuolia.
- CMA-tyyppi 3 (Kügelberg-Welanderin amyotrofia) voi näkyä missä tahansa iässä 2 vuoden iän jälkeen. Normaalisti kasvanut ja kehittynyt lapsi alkaa asteittain valittaa heikkoudesta, yleensä olkapäissä, käsivarret. Kun se etenee, hänen on vaikea ajaa, kävellä portaita, kyykky. Kaikki riippuu hoidosta - jotkut säilyttävät kykynsä liikkua itsenäisesti monta vuotta.
- CMA-tyyppi 4 (Kennedyn atrofia) esiintyy vain miespotilailla, koska sitä pidetään sidoksena X-kromosomiin. Ensimmäiset merkit ovat heikkous reiden lihasten alueella, ja kallon hermot vaikuttavat vähitellen. Sairaus etenee hitaasti.

hoito
Valitettavasti nykyään lääketiede ei voi tarjota menetelmiä ja keinoja hoitaa SMA: ta. Tällaisia menetelmiä ei ole. Jotta voidaan ylläpitää kehon toimintoja ja maksimoida ajanjakso, kunnes lapsi voi liikkua, lääkkeet, kuten Prozerin, Oksazil. Ne vähentävät asetyylikoliinia pilkkovien entsyymien aktiivisuutta, joka lähettää virityspulssin hermoston kuitujen läpi.
Suositellaan myös Sellaisten lääkkeiden järjestelmällinen käyttö, jotka lisäävät energia-aineenvaihduntaa solutasolla, B-ryhmän vitamiineja, nootrooppisia lääkkeitä sekä kalium- ja nikotiinihappovalmisteita.
Lapsi, jolla on SMA runsaasti proteiinia sisältävä ruokavalioViimeaikaiset tutkimukset ovat kuitenkin osoittaneet, että ruokavalion rooli on hieman liioiteltua - ei ole näyttöä siitä, että korkea proteiinipitoisuus ruoassa ainakin jollakin tavalla vaikuttaa taudin etenemisen nopeuteen.
Mutta kaloreiden pitäisi olla varovaisempia - lihasaktiivisuuden vähenemisen vuoksi lapsi voi nopeasti saada lisää kiloa.
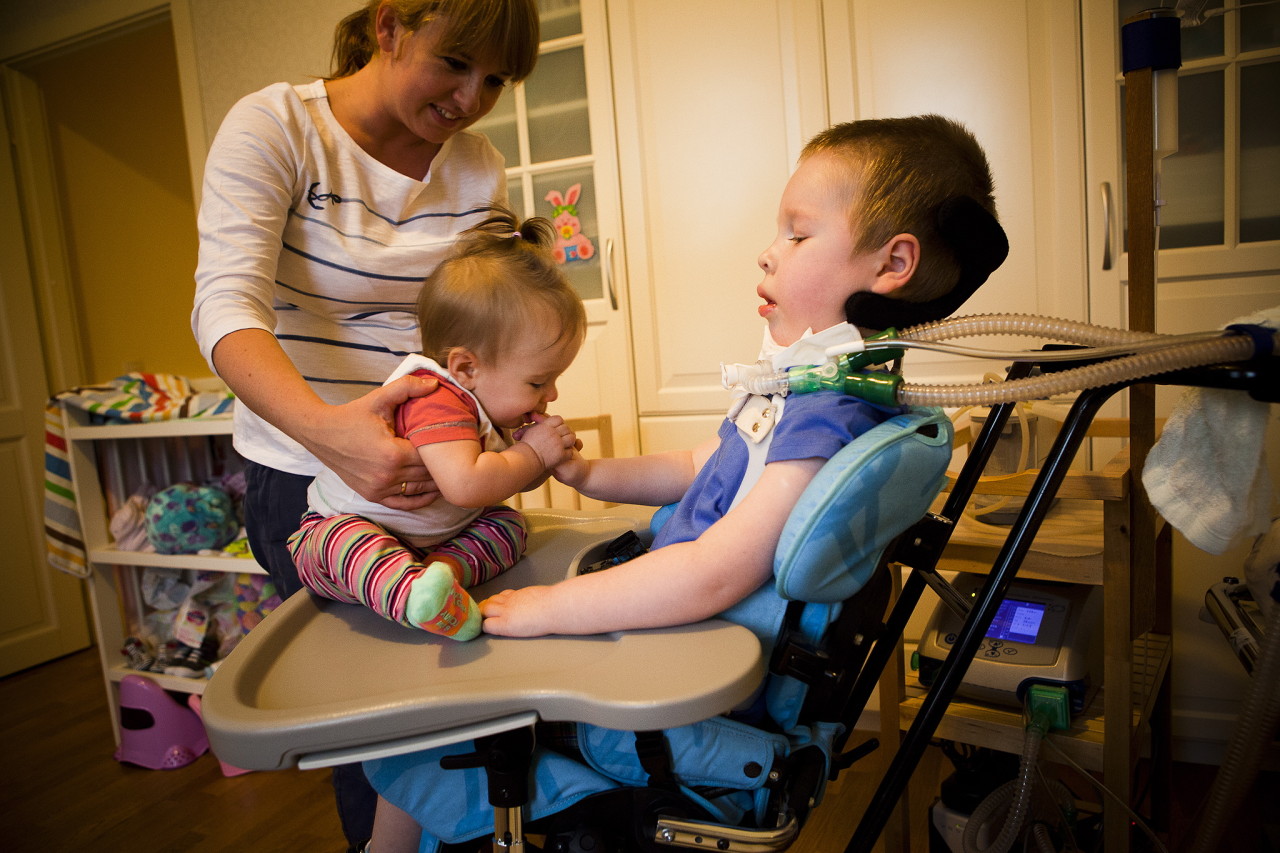
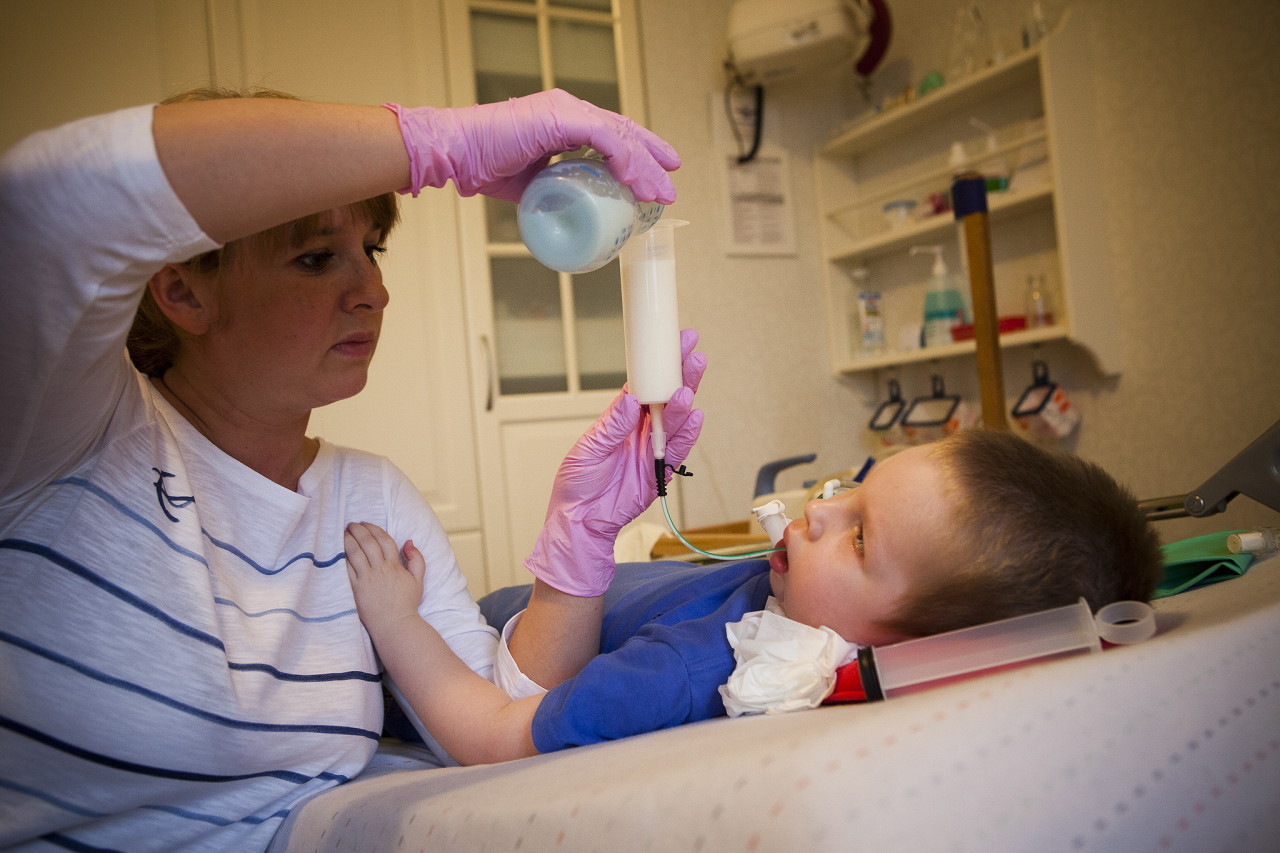
Auttaa pidentämään enemmän tai vähemmän täyttävän elämän aikaa terapeuttinen hieronta, UHF, elektroforeesi, hengitysharjoitukset hengityslihaksen ylläpitämiseksi, uinti. On suositeltavaa käyttää selkärangan tai rintakehän ortopedisia laitteita.

Lisätietoja taudista kertoo alla olevan videon asiantuntijalle.